第一节 免疫信息学源流
上个世纪中叶充满科学传奇。那不仅是个DNA双螺旋结构阐明、分子生物学产生与兴起的时代,也是系统论、控制论、信息论纷纷形成问世并引起全球广泛关注的时代,还是电子计算机走进人类社会并产生重要影响的时代。香农发表了《通信的数学理论》,奠定了他信息论之父的地位;维纳出版了专著《控制论-关于在动物和机器中控制和通讯的科学》;贝塔朗菲出版了系统论的代表作《一般系统理论:基础、发展与应用》。信息科学与计算机注定从一开始就与生命科学有着千丝万缕的联系。维纳专著的标题就有动物一词,贝塔朗菲本身就是生物学家。也就在那时,计算生物学(Computational Biology)、理论生物学(Theoretical Biology)悄然问世。进入上个世纪八十年代,随着人类基因组计划的酝酿与实施,数据驱动的生命科学研究开始横扫一切,生物信息学应运而生,日新月异,并不断渗透或应用到生命科学各学科甚至信息科学的一些前沿领域。
免疫学也不例外,生物信息学与免疫学的交叉融合形成了生物信息学的一个分支学科:免疫信息学(immunoinformatics)。俄亥俄州立大学已故外科与病理学系教授、移植免疫学家Charles G. Orosz在本世纪初首先创造了immuno-informatics一词,并将其定义为研究免疫系统产生、传递、处理及存储信息规律的学科(the study of the principles by which the immune system generates, posts, processes, and stores information)[1, 2]。或许人类偷懒的天性使然,在第239期的诺华基金研讨会(Novartis Foundation Symposium)*上,以色列魏茨曼科学院计算机科学与应用数学系的Lee A. Segel教授省掉该词中间的连字符,immunoinformatics这一词汇第一次出现在文献中[3]。在这篇题为“通过扩散信息网络的扩散反馈控制免疫系统”的文章中,Segel教授在文中写道[3]:“
免疫系统是研究生物信息处理的绝佳对象。由于免疫系统本身极为有趣,加上它在医学上的重要性,因此,虽然还有许多研究要做,但免疫系统的硬件相对已研究得较为清楚。在分子水平,显著的高频体细胞突变改变了遗传信息,使B细胞受体更具多样性。然而,更为吸引我的是在细胞水平,因为我相信,这一水平的规律,不仅决定免疫系统的行为,而且也能用于其它主要的生物系统,以及非生物的分布式自主系统。
脊椎动物拥有许多不同类型免疫细胞,总数可达1018个,却没有明显的老王(Boss)。不同类型的细胞群调动起来对付、攻击各种病原及毒株。此外,免疫系统也参与伤口愈合、组织重建等其它自身稳定任务。大量被称为细胞因子的信号分子指导着免疫系统。每种细胞因子具有几种功能,每种功能受几种细胞因子的影响。当配体与相应受体结合,通常分泌多种而非一种细胞因子。
这一超级复杂的分布式自主系统是如何确定做什么,何时做,做到何等程度的?我将从各个方面讨论这一问题,着重强调信息的作用。我尤其要论证基于细胞因子的扩散信息网络(diffuse informational network)所起的决定性作用。这样,我也就回应了Orosz关于免疫信息学(immunoinformatics)关键作用的建议。其中,免疫信息学被定义为研究免疫系统产生、传递、处理及存储信息的学科。”
2002年10月,诺华基金会资助下的史上首次免疫信息学研讨会在伦敦召开,来自实验免疫学、理论免疫学、生物信息学等各个领域学者畅所欲言[4]。在这次会议上(即第254期的诺华基金研讨会),会议主席德国教授Rammensee提出:免疫信息学可分为硬的、半软及软的三个领域,硬领域主要指免疫学相关核酸、多肽序列等数据及数据库工作,半软领域指处理免疫数据的方法及各种预测算法、软件等,软领域指免疫相关数学模型及其它各类理论免疫学研究[5]。此次研讨会迄今,免疫信息学已经过十余年发展,已经成为免疫学、生物信息学、计算机科学相结合的前沿领域。
广义地讲,当前免疫信息学可大体以下两大领域:
一是人工免疫系统(Artificial Immune System, AIS)及其应用[6, 7]。所谓人工免疫系统是指根据免疫系统的机理、特征、原理开发的人工智能算法与系统,如克隆选择算法、B细胞算法、阴性选择算法、树突状细胞算法等。人工免疫系统已应用到聚类分类、异常检测、信息安全、智能优化、图像处理、机器人控制、网络数据及文本挖掘等众多领域。该领域学者主要来自计算机与信息科学背景,模拟免疫机制,构建新的算法来解决各种科学及工程实践问题。该领域的研究内容从字面上看,非常贴近免疫信息学,源自免疫原理,落脚于信息方法。由于笔者对此领域没有深入研究,本章也不再赘述。
二是计算免疫学(Computational Immunology)及其应用。所有应用数学、计算机、人工智能、生物信息学方法来处理免疫学数据、分析免疫学问题的研究都属于计算免疫学,具体内容涵盖:理论免疫学(Theoretical Immunology)分析,免疫相关数学模型,各类免疫相关数据库构建,各种免疫相关预测算法、计算工具、网络服务及其应用如计算疫苗学(Computational Vaccinology)[8, 9]、反向疫苗学(Reverse Vaccinology)[10, 11]、计算机辅助疫苗设计[12]、计算机辅助抗体设计[13]等。一言以蔽之,用计算的方法解决免疫相关科学或工程实践问题就是计算免疫学,这也是当前免疫信息学的主流方向[14]。有趣的是,这一方向的工作从字面上看,用“信息免疫学”一词或许更为贴切,即源自信息学方法,落脚于免疫学问题。本章将结合笔者的科研工作,主要介绍计算免疫学资源及其应用。
第二节 免疫信息学资源
所谓免疫信息学资源,主要是指免疫相关的数据集、数据库、单机及网络程序等能够给用户解决相关问题提供便利与帮助的数据资料和软件工具。实验免疫学家、生物技术企业、咨询机构等作为这些资源的普通用户,可能并不太关心相关数据库如何构建、程序算法细节,但需要了解到哪儿去找特定数据,对于特定问题,什么软件好用等等。对于生物技术专业的同学——未来生命科学的研究者、生物技术的探索者、生物产业的创业者与经营者、政策的制定者与管理者,亦复如是。本节着重介绍开源、免费、共享、方便的网络免疫信息学资源。
1、免疫学数据库
在生物信息学研究中,构建专业的数据库、高质量的数据集是关键的基础性工作。没有相应的专业数据支持,后续研究工作无法开展。当然,有了数据仍不够,还必须有质控标准与措施来保证数据的质量,否则,生物信息学研究就是垃圾进、垃圾出。这就好比实验生物学研究,必需要有合适的模式生物及独到可靠的研究材料一样。
由于生物技术的飞速发展与信息技术的进步,海量的生物数据产生了,这就需要各种类型的数据库来存储、管理、检索各种类型数据,如核酸序列、蛋白序列、空间结构、表达谱芯片、网络与通路等。大量生物信息学数据库产生了,如GenBank[15]、UniProt[16]、MMDB[17]、KEGG[18]等。这些数据库当然也包含了部分免疫学相关数据,但构建专门的免疫学数据库对免疫学研究而言无疑更为专业与方便。自从1970年第一个免疫学数据库KABAT诞生以来,大批高度专一或涵盖广泛的免疫学数据库纷纷登场。本节我们重点介绍几个重要的门户型综合数据库及一些具有历史意义或特色鲜明的专一型数据库。
(1)免疫遗传信息的门户IMGT[19]
IMGT数据库最初由Marie-Paule Lefranc (Université Montpellier II, CNRS) 创建于1989年,目前已发展成为全球免疫遗传信息的门户。IMGT是“免疫遗传学(ImMunoGeneTics)”的缩写,目前包括7个子数据库。这些子数据库可分为序列数据库(IMGT/LIGM-DB、IMGT/MH-DB、IMGT/PRIMER-DB、IMGT/CLL-DB),基因数据库(IMGT/GENE-DB)、结构数据库(IMGT/3D structure-DB) 和单克隆抗体数据库(IMGT/mAb-DB)。IMGT/LIGM-DB是一个具有详细注释的人类和其他脊椎动物免疫球蛋白(IG)与T细胞受体(TCR)的序列数据库,目前收录了来自303个物种的16万多条序列。IMGT/MH-DB是一个人类主要组织相容性复合物(HLA)的专门数据库,其中包括世界卫生组织HLA系统命名委员会的官方序列;目前,该数据库服务器主机放在欧洲生物信息学研究所(EBI)。IMGT/PRIMER-DB是标准化的IG和TCR寡核苷酸探针或引物数据库;目前该数据库有来自11个物种的1864条记录。IMGT/PRIMER-DB提供的信息尤其适用于正常和病理情况下的IG和TCR的表达研究、单链抗体、噬菌体展示、基因芯片设计。IMGT/CLL-DB是一个新的数据库,主要收集来自慢性淋巴细胞性白血病患者的IG序列。IMGT/GENE-DB是人类、小鼠、大鼠、家兔IG和TCR基因的数据库;到目前为止,它收录了IG和TCR基因2893个、等位基因4139个。IMGT结构数据库收录IG、TCR、MHC空间结构及相关信息,目前有2686个记录。IMGT/ mAb-DB收录已上市及临床试验中的单克隆抗体、抗体-受体融合蛋白及其他免疫应用的融合蛋白,目前有单抗272个、抗体-受体融合蛋白18个,共计420个条目。此外,IMGT网站目前还提供了15个与数据库配套的在线分析工具。IMGT已广泛用于自身免疫、感染、肿瘤的相关医学研究、兽医学研究及抗体生物技术研究中。IMGT的所有资源都可通过其主页http://www.imgt.org免费使用。
(2)免疫表位信息的门户IEDB[20]
所谓表位(epitope)就是抗原中能被免疫细胞特异性识别的线性片段或空间构象性结构,是引起免疫应答和免疫反应的基本单位。表位在免疫学基础与应用研究中具有核心地位,是疫苗、抗体药物、肿瘤免疫、移植免疫、超敏反应的结构基础。IEDB是免疫表位数据库(Immune Epitope DataBase)的缩写,创建于2003年。IEDB虽然不是最早出现的表位数据库,但在NIAID的资助下,经过10年的发展,它已毫无疑问地发展成为全球免疫表位信息的门户。目前IEDB收录的表位及相关信息涵盖除肿瘤和HIV以外的99%已发表文献。文本挖掘程序自动扫描了2200万多篇PubMed文摘;大量专家对其中13000多篇文献(含7000多篇感染相关文献、1000多篇变态反应相关文献、约4000篇自身免疫相关文献、1000篇移植免疫相关文献)进行了人工归类、信息提取与注释。到笔者撰写此节为止,IEDB已提取文献14718篇,收录了人类及其它各种动物识别的多肽表位92407个,非多肽表位1831个。IEDB还包含了直接提交及来自FIMM、TopBank等一系列MHC配体数据库的数据,共有MHC配体(抗原肽)214704条。IEDB收录的表位不仅数量最多,而且质量最好,包含的表位相关的各种背景信息最为丰富,甚至连实验细节也不放过。此外,IEDB还提供了一些配套工具用于表位预测与分析[21]。所有IEDB数据及工具均可通过www.immuneepitope.org免费访问与使用。
(3)免疫多态性数据库IPD[22]
该数据库由安东尼.诺南研究所(Anthony Nolan Research Institute)#的HLA信息学小组与欧洲生物信息学研究所紧密合作,创建于2003年,旨在为研究免疫系统基因的多态性提供方便。IPD目前包括MHC、KIR、HPA、ESTDAB等四个子数据库。IPD-MHC数据库收录了大量物种的MHC序列。这些物种包括:家犬、狼、山狗,家猫,僧帽猴、狨猴、枭猴、松鼠猴、绢猴、叶猴、狒狒、猕猴、白眉猴以及其它猴类,倭黑猩猩、黑猩猩、长臂猿、大猩猩、猩猩等。ESTDAB是一个肿瘤细胞系的数据库。IPD最具特色的是KIR数据库和HPA数据库;KIR收录了人类杀伤细胞免疫球蛋白样受体(Killer-cell Immunoglobulin-like Receptors, KIR)共614个各等位基因及相应蛋白质的序列;HPA收录了人类同种异体血小板抗原数据。这些数据库均可通过http://www.ebi.ac.uk/ipd免费使用。
(4)HIV数据库
这是第一个面向病原体的数据库,收录获得性免疫缺陷病毒(HIV)的核酸序列、免疫表位、耐药相关突变及疫苗试验。其中,HIV分子免疫学数据库始建于1987年,由洛斯阿拉莫斯国家实验室开发,最初的目的只是提供一个已知HIV表位的全面列表,包括细胞毒性T细胞表位、辅助性T细胞表位和抗体结合位点。目前这一模式已推广到其他病原体,如丙型肝炎病毒(Hepatitis C Virus, HCV)和出血热病毒(Hemorrhagic Fever Viruses, HFV)。HIV数据库提供了大量分析工具,如表位比对(QuickAlign)、PeptGen、基序检索(Motif Scan)、序列定位(Sequence Locator)、ELF(Epitope Location Finder)等等。QuickAlign可用于比对表位、功能域或其它任何感兴趣的蛋白质区域。PeptGen可创建蛋白质的重叠肽图谱,有助于多肽设计与表位确定。基序检索工具可找出蛋白序列中具有某一指定HLA基因型、血清型或超型基序的亚序列。序列定位工具能给出用户序列相对于HIV参考序列(HXB2)或SIV参考序列(SIVMM239)的位置。整个项目受NIAID资助,所有数据及工具均可通过http://www.hiv.lanl.gov免费获得或使用。
(5)KABAT 数据库[23]
为了确定抗体序列中的抗原结合部位,著名免疫学家Elvin A Kabat(1914-2000)和他的研究小组于1970年创立了KABAT数据库;这也是第一个免疫学数据库。现在的KABAT数据库包括来自许多物种的IG、TCR、MHC及其它免疫相关分子的序列。一方面,新的序列仍在不断加入KABAT数据库;另一方面,该数据库相关信息分析工具也越来越多,这些工具包括关键词搜索、变异性分析和序列比对等。KABAT数据库及相应工具曾经可以从http://immuno.bme.nwu.edu免费获得;但现在只有付费注册才能使用。2003年以后,该数据库不再更新,第一个免疫学数据库就这样在不适宜的商业模式下成为僵尸数据库。
(6)SYFPEITHI数据库[24]
1999年,Hans-Georg Rammensee教授及其同事开发了一个关于MHC配体和基序的数据库—SYFPEITHI。该数据库的名字来源于第一个直接测序的天然MHC配体,即SYFPEITHI九肽;该配体洗脱自小鼠P815肿瘤细胞的H-2Kd分子。目前,SYFPEITHI数据库收录了来自人类和其他物种(如猿、牛、鸡、小鼠等)MHC-I 类和 II类配体的序列7000多条,基序200多种,所有的数据仅来源于文献。SYFPEITHI数据库工具可检索等位基因、基序、天然配体、T细胞表位、源蛋白质/物种及参考文献。数据库与EMBL和PubMed数据库有超链接。SYFPEITHI数据库提供了一个基于基序打分的表位预测界面,可预测人类及小鼠的多种MHC分子配体,通过www.syfpeithi.de免费在线使用。SYFPEITHI数据库的脱机版本,可在个人电脑或局域网内使用,但需付费(单机每年3000欧元)。又一个具有历史意义的数据库在商业化模式中迷失。
(7)未完待续
从半抗原小分子、佐剂、表位、抗原、变应原到抗体,但凡当前免疫学课本上有的,互联网往往已有相应数据库。例如:半抗原数据库(HaptenDB[25]),佐剂数据库(Vaxjo[26])表位数据库(Bcipep[27]、CED[28]),保护性抗原数据库(Protegen[29])、肿瘤抗原数据库(TANTIGEN、CIDB[30]),变应原及表位数据库(ALLERDB[31]、Farrp、InformAll[32, 33]、SDAP[34]),抗体数据库(SACS[35]、Abysis)等等,不一而足。从纸质版到电子版,从平面文件(flat file)到关系型数据库,从单机到网络,免疫学数据库经过几十年的发展,种类与数量越来越多,数据库的容量越来越大,结构也越来越复杂。除了这些数据库外,免疫学相关的测试数据集(Benchmarks)也是重要的资源。由于篇幅限制,我们不再列举免疫学数据库及数据集。好事者,可通过 Google等搜索引擎,Nucleic Acids Research、Database及其它专业期刊查询相关数据库与数据集。
2. 单机软件与网络程序
有了数据的支持,理论免疫学家就可以通过数学方法建立理论模型,进行模拟仿真;计算免疫学家则可在此基础上进行数据挖掘,建立新的算法,或用这些数据训练人工神经网络、支持向量机,建立新的预测模型并进行测试、评估与分析。这些模型、仿真、算法及预测器往往最终实现为单机或网络程序。如前所述,网络程序已经成为免疫信息学乃至生物信息学软件开发的大趋势,但出于数据安全保密等各种考虑,单机软件仍受一些公司与研究组的青睐。另外一个趋势是数据库自带的分析工具越来越多,数据库与网络程序的界限日渐模糊。此外,不少时候,解决一个免疫学问题往往需要一系列小的软件工具与步骤,由于这些小软件大都是现成的,因此只需要进行系统集成,形成合理的工作流程即可,这样的工作可通过诸如Accelrys公司的Pipeline Pilot等平台来自动化。由于免疫学相关网络程序不可胜数,本节不再具体罗列,而在免疫信息学的具体应用中部分提及。
第三节 免疫信息学的应用
免疫学不仅是研究分子识别与相互作用的基础学科,而且贴近应用。FDA近年批准进入市场销售或临床试验的药物中,新型抗体与新型疫苗屡拔头筹。由于抗体药物的巨大成功,以新型抗体、新型疫苗开发为核心的免疫学研究已经成为世界各国生物技术学术界与产业界全力追捧与投入的领域。与生物信息学相结合免疫信息学研究不仅可以解决一些分子识别的基本生物学问题,而且也必将为产业界提供新的工具。免疫信息学资源已在生命科学基础研究与应用开发研究中广泛应用。这里,笔者结合自己的研究工作进行部分介绍。
1. 表位预测
表位预测是免疫信息学研究的核心问题之一,也是抗体、疫苗、移植免疫、变态反应计算分析的基础。因此,发现一个重要的新表位和发现一个新基因一样,都蕴藏着巨大的财富。因此,从IT巨人微软到世界500强的生物医药公司罗氏,都有人在做表位预测研究。1999年,纳斯达克上市公司Epimmune(Nasdaq:EPMN)将其发现鉴定的一个可能用于乳癌、肺癌、结肠癌治疗的候选"先导表位"作价200万美元卖给了给美国Searle公司(世界500强的转基因寡头孟山都公司的制药部)。靠着它的另一项专利成果泛DR表位PADRE,Epimmune公司还从Elan公司、Pharmexa公司获得了不菲的非独占授权费,而后,Genencor公司更为此付出了6000万美元的独占性授权费;不仅如此,Epimmune公司今后还将享有上述3家公司所有PADRE相关产品的销售提成。
回到现实,如果我们将抗原比作一篇文章,那么表位就好比文章的关键词;而表位簇集区域就好比是摘要。根据表位特异性免疫应答的程度,可将抗原中的表位分为免疫优势表位、亚优势表位和隐性表位;根据表位对机体的影响,可分为保护性表位(免疫位)、致病性表位(变应位)、耐受性表位(耐受位);根据识别的免疫细胞,可分为B细胞表位、辅助性T细胞(Th)表位、细胞毒性T细胞(Tc)表位等。表位预测就好比对抗原这篇文章进行钩玄摘要。目前有关研究主要是B细胞表位、Th表位、Tc表位的预测上。
(1)B细胞表位预测
B细胞表位要被抗体识别,只能位于抗原表面。因此,早期的线性B细胞表位预测采用唯象方法(Phenomenological theory),通过计算蛋白亚序列的理化性质或二级结构,利用B细胞表位与上述理化特性或二级结构的相关性进行预测。例如,亲水的氨基酸更多位于液相面,而疏水的更多地深埋在抗原内核,因此可以计算蛋白质序列局部的亲水性来间接推断B细胞表位。对蛋白序列局部理化性质或二级结构倾向的理论计算大多依据相应的属性量表,这些量表可通过实验或统计分析得到。常用的量表如Janin可及性量表、Hopp和Woods亲水量表、Parker亲水量表(示例见图7-1)、Thornton突出指数量表、Welling抗原性量表等。30多年来,经典量表时有优化更新,对滑窗法的预测性能也有所改进。
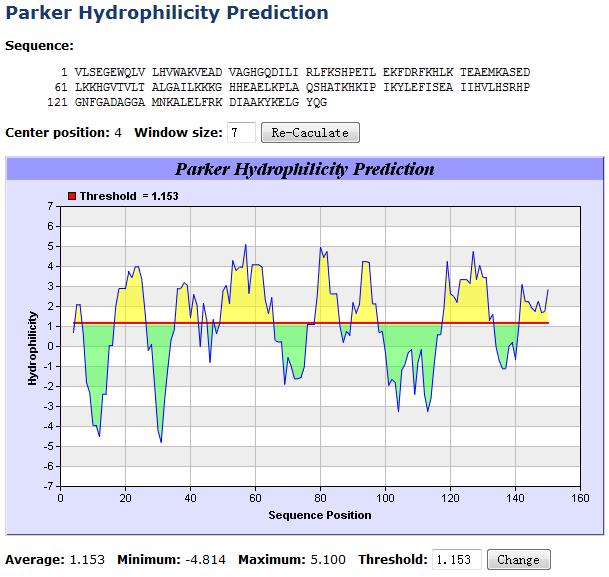
图7-1 基于Parker亲水量表的线性B细胞表位预测示例
复杂网络(Complex networks)已经成为近年自然科学乃至社会科学诸多学科领域研究的一个焦点与超级热点。现实世界中的很多系统都可以用复杂网络的模型来描述与研究,如互联网、集成电路、电力网、通讯网、铁路网、食物链、人际关系网等等。各种网络在数学上都可以用图(Graph)来表述。图论(Graph theory)源于18世纪大数学家欧拉(Leonhard Euler)对规则图(regular graph)的研究;20世纪50年代,两位匈牙利科学家Erdös和Rényi的研究使经典图论出现重大突破,确立了随机图理论(Random Graph Theory)。但不管是表征何种网络的图,它们都是由一些基本单元和它们之间的连接构成的;在图论中,我们通常把前者称为节点(nodes)、顶点(vertices)或点(point),把后者称为边(edges)、连接(links)或者线(lines)。基于图论的系统生物学研究已经取得了许多重大成果,食物链网络、大脑皮层神经元网络、代谢网络、基因调节网络等许多生物系统的复杂网络已被证实具有小世界(small-world)或/和标度无关(scale-free)特性。在基因调节网络、代谢网络的研究中,蛋白质(或酶)被简单地看作为网络节点。但是,当把研究聚焦于蛋白质本身或其部分片段的时候,例如对蛋白质抗原,我们同样可以把抗原看作一个复杂网络,一个大的“图”。它的节点是氨基酸残基,它的边是氨基酸残基之间在序列上与空间结构上的联系(如图7-2)。
图7-2:蛋白质抗原氨基酸残基网络示意图(家兔子宫珠蛋白: 1UTG)
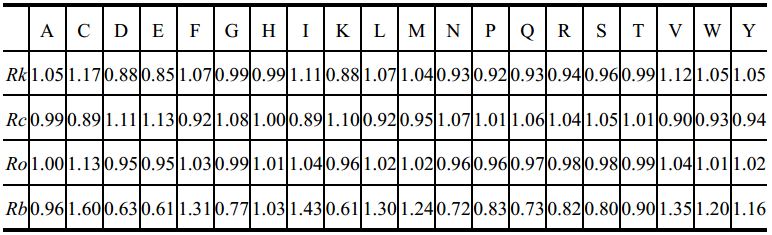
我们从PDB数据库中筛选出无冗于、高分辨率的代表性晶体结构640个,以氨基酸残基为节点,残基之间序列上的联系(实质是肽键)或空间上的联系(实质是二硫键等其它共价键或氢键、疏水相互作用、范德华力等非共价相互作用)为边,构建了640个抗原的氨基酸残基网络。计算这些网络中每个氨基酸残基的连接度(Connectivity)、聚类系数(Clustering coefficient)、完形度(Closeness)、间度(Betweeness)等复杂网络参数,统计推演出基于复杂网络的4个新量表(见表7-1),即相对连接度(Rk)、相对聚类系数(Rc)、相对完形度(Ro)、相对连接度(Rb)。
表7-1 基于抗原氨基酸残基网络拓扑性质的新属性量表[36]
我们把上述新量表用于基于滑动窗的B细胞表位残基预测。结果显示,在5个测试数据集中,相对连接度(Rk)表现稳定优越,在注释最完整、质量最佳的HEL数据集中,相对连接度的ROC曲线下面积最大,提示预测性能显著优于Parker亲水性(Ph)及Levitt指数(Li)等两个已知最好的B细胞表位预测量表(见图7-3)[37]。因为相对连接度反应了一个氨基酸残基在网络中相邻氨基酸残基数目的倾向。偏爱在抗原内核的,邻居一般较多,在表面的,邻居相对较少。因此,用相对连接度来预测B细胞表位能有如此表现也就不奇怪了。
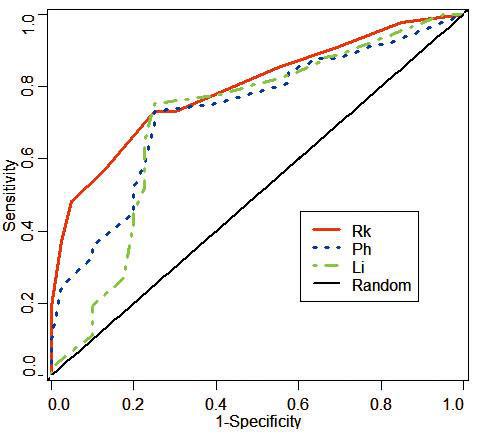
图7-3相对连接度、Parker亲水性及Levitt指数预测性能比较
专门的线性B细胞表位预测工具有PREDITOP、ADEPT、PEOPLE、BepiPred[38]等;一些综合性序列分析软件,如OMIGA、UWGCG、ANTHEPROT等,也包含了的线性B细胞表位预测功能。线性B细胞表位预测往往是根据蛋白质抗原的氨基酸序列,预测其上可能的B细胞表位区段。由于B细胞表位在没有其对应抗体时是不能确定的,它的存在取决于相应抗体的存在,所以,从学术的角度,目前的线性B细胞表位预测似乎是个伪命题。但就是这个命题却是目前大部分抗体公司急需解决,有着重大现实需求的。例如,根据预测结果,用从天然蛋白中筛选出的合成肽段来免疫动物,以期获得的抗多肽抗体能与天然蛋白本身发生交叉反应。这具有重要的实用价值,相应抗体既可用于亲和层析,达到分离纯化完整蛋白抗原的目的,又可用于免疫组化或其它免疫反应而有助于基础研究与临床诊断,可以开发为科研甚至临床诊断试剂。当前,人工神经网络、支持向量机等机器学习方法已用于线性B细胞表位预测,但性能仍不令人满意。此外,基于噬菌体展示或抗原空间结构的构象表位预测近年来也广受关注。前者我们将在后文中单独论述;后者包括Java语言编写的基于随机森林的单机程序Bpredictor[39],网络程序CEP[40]、DiscoTope[41]及SEPPA[42]等。
(2)T细胞表位预测
T细胞表位的预测研究肇始于对Th表位的预测。早期的Th表位预测明显受线性B细胞表位预测思路的影响,不少研究试图从实验证实的Th表位中找出它们在理化特性或二级结构上的共同特征并在一定程度上获得了成功,第一个Th表位预测程序AMPHI就是早期研究的代表作品。二十世纪80年代末90年代初, MHC-I类分子晶体结构的阐明和多种Tc表位基序的发现使Tc表位预测研究率先取得突破。这带动了MHC- II类分子晶体结构与各种Th表位基序的揭示,使Th表位预测摆脱了B细胞表位预测思路的影响。目前,打分矩阵、隐马尔可夫模型(Hidden Markov Model,HMM)、比较分子力场分析(comparative molecular field analysis, CoMFA)、比较分子相似性指数分析(comparative molecular silmilarity indices analysis, CoMSIA)、人工神经网络(artificial neural network , ANN)、支持向量机(Support Vector Machine, SVM)等各种方法都已用于T细胞表位预测。相关单机及网络程序包括AMPHI、Tepitope、TSites、EpiMer、EpiMatrix、BIMAS、MHCPred、NetMHC、NetMHCpan、NetMHCcons、NetCTL、NetCTLpan、NetMHCII、NetMHCIIpan、MetaMHC[43]、TEPITOPEpan等。这个领域非常热闹,于是该领域元老Brusic教授于2009年组织了首届机器学习在免疫学中的应用——HLA-I类配体预测(MLI)大赛[44]。行笔至此,2012伦敦奥运与第二届MLI大赛正如火如荼,发扬奥运精神,我们也参与其中。希望有兴趣的同学今后能加入我们。
虽然对Tc表位预测的探索起步最晚,但进展最快,研究最深入,预测最成功,尤其是基于支持向量机的方法。目前,对Tc表位的预测,研究内容已经不只限于对MHC-I类分子结合及其结合能力的预测,而且还拓展到对候选Tc表位自然产生可能性及其转运效率的预测,即对蛋白酶体酶切位点及抗原处理相关转运蛋白(transporters associated with antigen processing, TAP)的转运进行预测,范围涵盖整个抗原处理与递呈过程,相关程序如FragPredict、PAProC、NetChop等。此外,近些年的研究越来越重视杂合性T细胞表位、超型表位及表位簇集区域的预测。
2. 噬菌体展示
(1)噬菌体与噬菌体展示
噬菌体(phage)是一类感染细菌的病毒。有些噬菌体,如丝状噬菌体M13、fd、f1等,是非常好的表达载体[45]。通过基因工程方法,可以将外源基因片段或随机DNA序列插入噬菌体基因III或VIII中,表达后形成的融合蛋白仍能够自主装配成噬菌体外壳并将外源蛋白或多肽展示在噬菌体颗粒表面(见图7-4)。
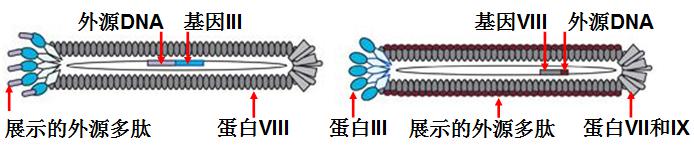
图7-4:通过基因III或VIII展示外源序列的M13噬菌体示意图
噬菌体易于培养,增殖迅速。通过随机PCR等分子生物学技术,可以很方便地构建多样性从数百万到数百亿的噬菌体文库(phage library)[46]。这就使得生物学家可以像钓鱼一样,从“鱼塘”(噬菌体文库)里高效地钓到能与“鱼饵(bait)”结合的展示了特定多肽的噬菌体。通常,用来筛选噬菌体文库的物质被称为靶位(target);靶位的天然配体称为模板(template)。如图7-5所示,使用靶位(如细胞、抗体、受体、酶、化合物、半导体材料等),经过几轮结合、洗脱、增殖的循环淘选(panning),通常能快速从噬菌体文库中筛出一系列可能与靶位特异性结合的噬菌体;通过DNA测序能快捷、可靠地推导出它们所展示外源多肽的氨基酸序列。这些能与靶位结合的外源多肽,通常模拟了相应模板的特定位点,因而被称为模拟肽(mimotope);而上述获得模拟肽的分子生物学实验流程与技术则被称为生物淘选(biopanning)或噬菌体展示(phage display)[47]。
噬菌体展示技术一经问世便迅速发展,并在现代分子生物医学研究中广泛应用。在基础研究中,利用噬菌体展示技术可以预测出表位[48]、其他蛋白质相互作用位点[49]与网络[50, 51],确定药物作用的靶标蛋白;在应用研究中,噬菌体展示技术可用于开发器官靶向试剂[52]、新诊断试剂[53]、新生物技术药物[54]和疫苗[55]。尤其值得一提的是,噬菌体展示技术在基于新型生物材料的新能源如生物锂电池[56]、氢能源[57]等研究中大放异彩,美国总统奥巴马曾因此专程参观了麻省理工学院的相关实验室。
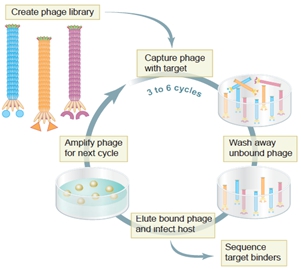
图7-5 噬菌体展示技术示意图(Science 2002)[58]
(2)噬菌体展示数据中的信号与噪声
在噬菌体展示实验的结果中,信号是研究者想要得到的模拟肽序列,也就是能特异性结合靶位的多肽。但是,由于噬菌体展示技术本身固有的原因,在淘选得到模拟肽的同时,也将不可避免地得到一些噪声序列,也就是所谓的靶位无关多肽(target-unrelated peptides, TUP)[59, 60]。这些噪声序列可分为两类。一类是选择相关TUP (Selection-related TUP, SrTUP),专指发生在噬菌体的结合选择环节,能够结合筛选体系中某种成分但并非与靶位特定位点结合的噬菌体展示多肽。由于靶位本身及淘选体系的复杂性,文库中的噬菌体除了与研究者期待的靶位位点结合外,还可能与淘选体系中的固相基质(如塑料和磁珠)、检测放大分子(如链亲和素、蛋白A)、封闭试剂(如牛血清白蛋白)、污染分子(如胎牛血清中的牛免疫球蛋白、大肠杆菌内毒素)及靶位的其它位点结合。另一类是增殖相关TUP (Propagation-related TUP, PrTUP),专指发生在噬菌体增殖环节,不是因为能与靶位结合,而是因为具有更快、更强的增殖能力而进入实验结果里的多肽[61]。与选择相关TUP一样,增殖相关TUP也极其常见,尤其多见于最为常用的基于M13噬菌体的Ph.D.系列商业化文库[62]。基于fd-tet噬菌体的文库增加了抵抗增殖相关噪声的设计;即便如此,增殖相关噪声序列仍不时出现在噬菌体展示实验结果中[61]。实验结果显示,增殖相关TUP不仅可能混入噬菌体展示的结果中,有的时候甚至会主宰噬菌体展示数据[62]。计算机模拟结果显示,噬菌体增殖能力的差异即便微小到10%,经过几轮增殖后也能引起噬菌体文库克隆丰度的极显著差异[63]。因此,噬菌体展示的实验结果往往是模拟肽与靶位无关多肽的混合物。
如果不加区分,将噪声作为信号,也就是把噬菌体展示实验结果中的靶位无关多肽当作模拟肽,不言而喻,其研究结果往往是误导的和悲剧的。令人遗憾的是,这种情况相当常见。例如,全球有几十个研究组都报道过一种序列为SVSVGMKPSPRP的神奇“模拟肽”。汇总各研究组的实验结果,提示它可能与40多种不同的靶位结合[64, 65]。这些靶位包括:多种器官与组织(如头发、皮肤、小鼠肿瘤血管、小鼠胚胎),多种细胞(如神经元、前列腺癌细胞、肝癌细胞、小鼠卵细胞、金黄色葡萄球菌),RNA,DNA,多种抗体,多种酶(如葡萄糖氧化酶、乙酰胆碱酯酶),多种蛋白(如神经生长因子、艾滋病毒Vif蛋白),多种多肽及各种材料(如磷脂酰丝氨酸脂质体、脑膜炎球菌脂多糖、羟基磷灰石、聚四氟乙烯、墨水染料、单壁碳纳米管、钴纳米颗粒、铂铁合金及磷化铟、砷化镓、氮化镓等半导体材料)结合。难道SVSVGMKPSPRP是无所不能的万能胶水?实际上,上述多数实验里它只是一条增殖相关的TUP。然而,具有讽刺意味的是,相当多的实验小组把噪声作为信号,最终得出了一些错误的结论还如获至宝。
令人欣慰的是,在噬菌体展示领域,无论是实验研究者还是理论研究者,都已经开始注意到实验数据中的噪声问题。在进行后续研究之前,很有必要预先减少或尽量消除实验数据中的靶位无关多肽,这已逐渐成为本领域研究者的共识。因此,靶位无关多肽的研究已经成为近年来噬菌体展示领域实验与生物信息学研究的热点。通过采取消减淘选(subtractive panning)、提高靶位结合选择的严谨度、减少淘选次数等各种实验措施,能在一定程度上减少选择相关及增殖相关噪声。但是,仅靠实验本身的改进并不能彻底消除靶位无关多肽,这是由噬菌体展示内在的结合选择与感染增殖环节所决定的[63]。因此,借助信息学手段来检测噪声序列,进而对噬菌体展示数据进行计算纯化等一系列预处理,不失为省时、省力、省钱的上佳之选。
(3)噬菌体展示数据预处理研究进展
目前,噬菌体展示数据的预处理方法大体可分为三类。一是是基于信息论的方法,二是基于TUP序列特征的方法,三是基于数据库搜索比对的方法[66]。
早在2004年,Mandava等就根据香农的信息论提出了用信息含量(Information content)这一指标来衡量噬菌体展示结果中每条多肽是信号还是噪声[67]。对于噬菌体展示实验结果中任意一条多肽序列X1X2…XN,其出现在初始文库中的概率P = P1×P2×…×PN,其中PN代表着初始文库中某种氨基酸出现在第N位的频率。多肽的信息含量INFO =﹣㏑(P)。Mandava等所谓的信息含量类似于信息论中的信息熵(information entropy)。Mandava等认为,初始文库中越罕见的多肽,信息含量越高,经过几轮淘选还能出现在结果中,这就越不可能是随机事件,这样的多肽应该是有意义的信号;反之,越是具有增殖优势的噬菌体,在初始库中就越常见,信息含量也就越低,如果出现在结果中则很可能是噪声序列。根据上述假设,他们开发了INFO程序[63]。
2010年,我们研究组根据Menendez等的综述,总结了当时所有已知靶位无关多肽的序列特征,编写了一个基于TUP序列特征的靶位无关多肽检测、报告与滤除程序(SAROTUP,http://immunet.cn/sarotup)[68]。我们的测试结果显示,给噬菌体展示实验数据增加一个基于TUP序列特征的预处理环节,不仅能极大地提高Pepsurf、Mapitope等程序预测蛋白质相互作用位点的性能,还有助于基于噬菌体展示的候选疫苗筛选[68]。
为了检测不具备已知TUP序列特征的靶位无关多肽,我们接着又构建了一个噬菌体展示实验结果的数据库(MimoDB,http://immunet.cn/mimodb),收集了全球各研究组利用噬菌体展示淘选随机文库的实验结果[64]。该数据库每季度更新,笔者成文时版本有数据1956套,多肽序列16500条,是目前世界上最大、最全的噬菌体展示实验结果数据库。去年底,我们又开发了MimoSearch、MimoBlast等几个基于该噬菌体展示数据库的小工具。通过MimoSearch小工具搜索数据库,可以查看实验者提交的多肽序列是否与其它已发表的结果完全相同。如果多个研究组在淘选实验中使用的靶位不同却又得到了完全相同的多肽序列,那么相应多肽极可能是噪声序列,即便它很可能不具备任何已知的噪声序列特征。通过MimoBlast小工具可对MimoDB进行全数据库序列搜索比对,不仅可以找到与实验者提交多肽序列不完全相同但高度相似的已发表结果,从而提示靶位无关多肽,还能找出与已知TUP高度相似的多肽从而推导新的TUP序列特征[65]。目前,这些基于数据库搜索比对的工具也已集成到了SAROTUP程序的2.0版中。
(4)基于噬菌体展示的表位预测[49]
由于模拟肽和抗原上的天然表位能与同一抗体结合,所以,通常认为,模拟肽及其所模拟的天然表位应该具有相似的物理化学性质和空间关系。这是目前所有基于噬菌体展示的表位预测方法共同的假设与前提。在这个假设与前提下,通过比对模拟肽序列(或一套模拟肽的共同序列)与抗原序列,抗原序列中与模拟肽序列匹配(一致或高度相似)的部分可能就是相应的天然表位。这种情况已得到不少实验结果的验证。但是,在针对蛋白质抗原的体液免疫中,所产生的抗体约有90%所识别的都是空间构象性表位;或者说与抗体结合的绝大多数天然表位在序列上都是不连续。因此,基于噬菌体展示的表位预测,在绝大多数情况下,模拟肽与抗原在一级结构上没有或者仅有非常低的相似性。因此,如何把模拟肽合理映射到抗原的空间结构上或分散的抗原序列中,就成为基于噬菌体展示的表位预测研究的核心问题。目前,相关算法、流程、单机及网络程序包括PEPTIDE、FINDMAP、EPIMAP、SiteLight、Mapitope、RELIC、3DEX、MIMOX、MIMOP、PepSurf、Pepitope、Pep-3D-Search、EpiSearch、MimoPro、LocaPep、PepMapper等[66]。
(5)噬菌体展示免疫信息学研究感悟
回顾既往7年我们在噬菌体展示领域的免疫信息学研究,我们做了数据集、数据库、数据预处理软件、数据解析预测软件,我们深深感受到了计算的力量。仅以我们的噬菌体展示实验结果数据库而言,这让我们充分体会到为什么Rammensee教授要将之称为hard了。数据的核实、提取及无休止的更正与更新,让数据库工作真的非常艰苦hard。当然,Rammensee教授的原意是指这些直接来源的实验结果的数据是如何hard,如何可靠;而建立在数据之上的算法与预测软件,那产出就软了,因为是计算预测结果,但好歹还可通过实验验证,所以就算个半软吧;至于无法实验验证的数学模型与仿真,那就全软了。这种认识,笼罩整个生物医学界,那就是:实验最可靠,实验是金标准;生物信息学,奇技淫巧罢了,计算的结果靠不住。但是,当我们搜集所有噬菌体展示实验结果到MimoDB数据库中时,我遇到了实验研究的各种怪状;当所有噬菌体展示实验结果通过MimoDB平台可以相互对照后,我们发现,实验结果也不都是可靠的,实验结果也不都是金标准,以子之矛、攻子之盾的地方实在太多。意外地,MimoDB可以成为一个噬菌体展示领域的循证生物学分析平台,可以帮助实验研究者消除实验结果中的噪声,这就是数据库的力量。
3. 在抗体研究中的应用
目前,有很多单机及网络程序可用于抗体的基础与应用研究的方方面面[13]。例如,使用Abnum可按Kabat、Chothia等三种方式给抗体序列定位编号,从而确定CDR区[69];使用SUBIM确定抗体重链、轻链的亚类[70];使用AbCheck检查提交的抗体序列中是否可能存在克隆伪迹(cloning artifacts*)或测序错误[71];使用SHAB评估抗体的人源化程度[72];使用PAPS预测抗体重链与轻链堆叠角度(VH/VL packing angle)[73];使用AbM、WAM[74]、SWISS-MODEL [75]、PIGS[76]、RosettaAntibody[77]等建立抗体的三维结构模型。此外,计算方法也可用于抗体亲和力成熟、防止凝聚、延长半衰期的设计等各个方面[13]。
免疫信息学已经极大地改变了单克隆抗体产业。目前,抗体测序已成常规,而一旦有了抗体序列,就可以通过序列分析确定其重链、轻链类型而无需实验确认;通过同源建模、分子对接及动力学等分析更能推断其相应抗原的大致情况。我们与华西医大合作,对他们通过杂交瘤技术获得的抗苏丹红I单抗进行了免疫信息学研究,通过从头计算与同源建模相结合的方式,获得了其空间结构的理论模型(见图7-6)。该图左侧为抗体轻链,其绿色、水色、蓝色、紫色部分对应LFR、L1、L2、L3;右侧为抗体重链,其红色、水色、蓝色、紫色段分别对应HFR、H1、H2、H3。正中凹洞为抗原结合位点,凹洞底部主要由重链骨架区形成,侧壁主要由L3和H3形成。整个图像由通过PMV软件以分子表面的方式显示,其中分子表面用MSMS软件计算。
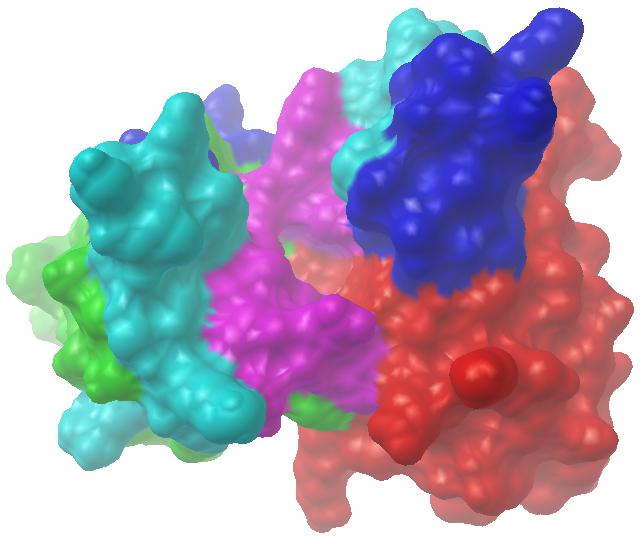
图7-6 抗苏丹红I单抗可变区空间结构理论模型
我们在剑桥结构数据库(Cambridge Structural Database, CSD)中查询到的苏丹红I的晶体结构实验数据共4条,没有苏丹红II、III、IV晶体结构的实验数据。使用Gaussian软件,计算出苏丹红I-IV的空间结构。将4条实验验数据与理论计算的苏丹红I结构进行叠合,发现实验数据之间RMSD差别竟然比与理论结果之间的差异还大,说明计算结果精确可靠。用Autodock4.1将理论计算的苏丹红I-IV结构与抗苏丹红I单抗模型进行了盲对接与定位对接,得到了苏丹红I(见图7-7)到苏丹红IV与抗苏丹红I单抗可能的结合模式。颇为有趣的是,苏丹红I、II与抗苏丹红I单抗的抗原结合位点对接结合时构象相似:“2-萘酚”(naphthalen-2-ol)基团在内,其余部分朝外;苏丹红III、IV与抗苏丹红I单抗的抗原结合位点对接结合时构象相似:2-萘酚”(naphthalen-2-ol)基团在外,其余部分在内。

图7-7 抗苏丹红I单抗-苏丹红I相互作用示意图
在上图中,抗体轻链绿色,重链橙色,均按卡通模式显示。根据CCP4软件包计算结果,苏丹红I在抗原结合袋中与轻链的H34、Q89、T97(绿字标出)及重链的V37、W47、E50、V97、K98、W103等主要通过范德华力相互作用,图中仅显示了这些残基中直接与苏丹红有范德华相互作用的重原子。此外,苏丹红I的N13原子与轻链T97残基的OG1之间距离2.69 Å,形成氢键(黄色虚线)。抗苏丹红I单抗与苏丹红II的相互作用与抗苏丹红I单抗-苏丹红I的相互作用非常相似,苏丹红II的N13原子与轻链T97残基的OG1之间距离2.67 Å,氢键更强,不再另图显示。
我们使用了SplitPocket[78]计算了抗苏丹红I单抗的抗原结合袋的体积以及苏丹红I、II、III、IV分子的体积。在抗苏丹红I单抗所有袋结构中,最大的袋位于抗原结合位点,我们将其称为抗原结合袋。它的尺寸为 5.23 Å × 6.64 Å × 10.36 Å(宽×厚×深度),体积为359.56 Å3;而苏丹红I、II、III、IV分子的体积分别为228.28 Å3、261.12 Å3、323.76 Å3、356.56 Å3。由于已有的研究显示[79]:结合袋往往都要比配体大得多,配体极少能够占据整个结合袋,所以,以上计算结果显示,抗苏丹红I单抗的抗原结合袋容纳结合苏丹红I、II分子比较容易,而要容纳苏丹红III则有些困难,苏丹红IV则几无可能。这些结果,与对接结合能数据也是较为一致的。总之,我们免疫信息学的计算分析提示:苏丹红I、II能与抗苏丹红I单抗的抗原结合位点结合,强度相近;苏丹红III可能有较弱的结合,而苏丹红IV不大可能与抗苏丹红I单抗结合。我们的结果返回华西后与他们实验的结果不尽一致!他们的实验结果显示,抗苏丹红I单抗与苏丹红I亲和力最高,其次苏丹红III,与苏丹红II结合较弱,不结合苏丹红IV。是实验错了还是我们的计算不对?后来,我们的合作者反复研究,发现当初结合实验时未考虑苏丹红I-IV之间溶解度的差异,纠正后的实验结果与我们计算的一致。计算的力量,由此可见一斑!我们也进一步仔细阅读他们的专利,发现免疫用的半抗原是苏丹红I修饰物(苏丹红I号-3-丙酸,Sudan1-C3)而并非苏丹红I本身。Sudan1-C3与OVA、BSA等偶联后,除了桥联的羧基碳原子外,其结构比苏丹红I多了2个碳原子,而与苏丹红II 的一个甲基碳原子正好重叠,另一个碳原子位置不同。因此,Sudan1-C3与苏丹红I、II很相近,推测引起的单克隆抗体能与苏丹红I、II发生较强交叉反应。
我们也曾对抗CD147的美妥昔单抗(metuximab)进行过同源建模,其理论模型见图7-8。该图左侧为轻链,其绿色、水色、蓝色、紫色部分对应轻链骨架区(LFR)、轻链互补决定区1、2、3(L1、L2、L3);右侧为重链,其红色、水色、蓝色、紫色段分别对应HFR、H1、H2、H3。美妥昔单抗的抗原结合位点呈两端开放的扁槽状,L3和H3构成槽底,L1、L2构成左槽壁,H1、H2构成右槽壁,LFR2区的F49及HFR3区的R94残基也参与了形成美妥昔单抗的抗原结合位点。从图中不难看出,针对蛋白质抗原的抗体与先前针对化学小分子的抗体,它们所形成的抗原结合位点形态迥然不同。对于任一抗体,通过建模后对抗原结合位点形态的观察,似乎就能大概知道其所针对抗原的大致类型。计算之神奇,叹为观止矣!
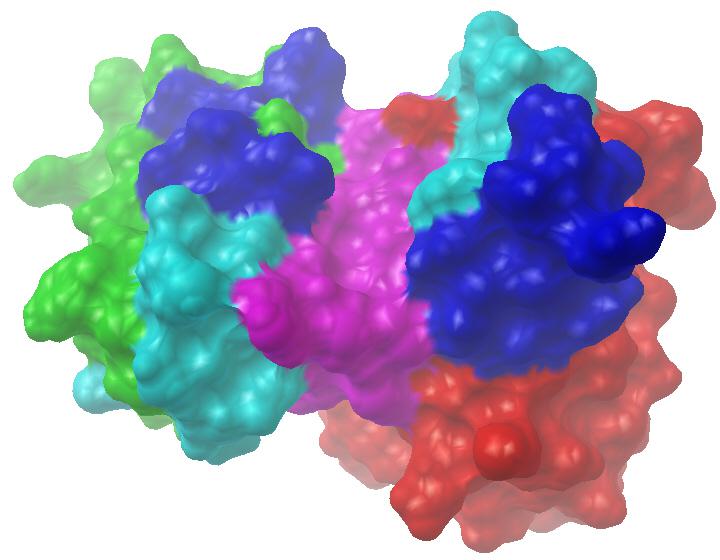
图7-8 美妥昔单抗三维空间理论模型
4. 在疫苗研究中的应用
疫苗极大地促进了人类健康事业的发展。但是,人类的生命与财产仍面临巨大的威胁。这些威胁主要体现在以下五个方面。其一,是新的传染病病原不断出现,如埃博拉病毒、人类获得性免疫缺陷病毒(HIV)、新的传染性肝炎病毒、疯牛病病毒及SARS病毒等。其二,是一些为人熟知的人畜致病微生物不断产生新的变种或耐药性并再次成为人类生命的杀手与财富的终结者,如大肠杆菌O157、结核分枝杆菌、口蹄疫病毒、禽流感病毒等。其三,是911后整个世界面临着日益严重的生物恐怖主义威胁,如911后的炭疽恐慌。其四,是大量疑难疾病急需新的治疗手段,如癌症的免疫治疗。其五,是对药品及疫苗本身生物安全性的担忧,如传统疫苗通常使用某种疾病的减毒或灭活的致病原,但这仍可能造成疾病传播或中。正因为如此,人类社会才迫切需要更快地拥有更多、更安全、更可靠的新的疫苗产品。
就以疫苗最经典的应用领域——传染病预防为例,目前已明确的致病微生物超过400中,但只针对其中30多种开发出了上市的疫苗。因此,仍然大量传染病预防疫苗仍亟待开发。目前的疫苗研发已从传统的预防性疫苗发展到治疗性疫苗,适用范围从原来单纯的传染病预防发展到对过敏性疾病、自身免疫性疾病、器官移植性疾病、计划生育、免疫去势、不孕不育症、老年痴呆、肿瘤防治、戒烟戒毒等各个方面。随着人类基因组计划的完成与大量病原微生物基因组的阐明,人类已进入从基因组到候选保护性抗原到保护性表位到个体化定制疫苗的时代,免疫信息学在其中发挥着重要的助推作用。
例如,在表位疫苗设计方面,EpiVax公司设计了iVAX工具包。iVAX是一套根据蛋白抗原序列,设计表位疫苗的免疫信息学工具,包括Conservatrix、EpiMatrix、ClustiMer、BlastiMer、EpiAssembler、Aggregatrix和VaccineCAD等程序。其中,Conservatrix评估九肽片段在同一病原不同毒株之间的保守性;EpiMatrix评估多肽片段的HLA-I、II分子结合能力;ClustiMer根据EpiMatrix输出确定富含T细胞表位多肽群;BlastiMer评估这些多肽群与人类基因组间的相似性,相似的序列要么耐受难以引起免疫应答,要么造成自身免疫病,因此需去掉;EpiAssembler将保守、特异且富含T细胞表位的多肽群合起来;Aggregatrix确定覆盖最多HLA型别,最多毒株病原的最小一套表位;VaccineCAD将候选表位多肽合理串接,避免因多肽连接形成新的表位。
疫苗研究在线信息网(Vaccine Investigation and OnLine Information Network, VIOLIN)也提供了大量疫苗研发相关信息,包括疫苗相关的各种数据库及程序[80]。其中,Vaxign是一个集成的反向疫苗学在线软件[81]。它包括两个部分:一是可直接查询预先已预测好的结果;二是对提交的新基因组或蛋白序列进行实时交互分析预测。Vaxign集成了一系列软件,包括:(1)确定蛋白抗原亚细胞定位的PSORTb,分泌或病原外膜蛋白可能是理想的候选靶标,而胞浆及内膜蛋白则不理想;(2)预测蛋白质跨膜方式的TMHMM,跨膜超过1次的蛋白难以克隆、表达、纯化,不适宜作为重组疫苗的抗原;(3)预测粘附及粘附样分子的SPAAN,粘附样分子的通常是理想的疫苗靶标;(4)预测MHC-I及II结合能力的Vaxitope,此软件由He研究组自己开发;(5)分析序列相似性的BLAST,与宿主高度相似的序列要么耐受要么导致自身免疫,不是理想的靶标。
最后,举一个实际案例。脑膜炎奈瑟菌是流行性脑脊髓膜炎(简称流脑)的病原菌。根据其荚膜多糖抗原的差异,可将脑膜炎奈瑟菌分为至少13个血清型,其中致病的主要是A、B、C、Y及W-135血清型。迄今,A、C、Y及W-135血清型的纯化多糖疫苗已使用了多年,取得了较好的预防效果;但到目前为止仍然未能开发出针对脑膜炎奈瑟菌B血清型(MenB)的疫苗。原因主要是MenB荚膜多糖一段与人唾液酸一致,其荚膜多糖不宜作为疫苗,而主要的外膜蛋白PorA变异度大,免疫后只能引起毒株特异性保护,不能预防所有MenB感染(见图7-9)。这样,MenB导致的流脑愈发常见,已占美国流脑的1/3,欧洲流脑的45%-80%以上,成为严重的健康威胁。2000年,MenB-MC58株的全基因组测序完成[82],生物信息学方法立刻用于预测其基因,免疫信息学方法立刻用于预测、筛选保护性抗原[83],预测与实验相结合,很快确定了在外膜上含量相对较少、但更加保守的几个保护性抗原如H因子结合蛋白(factor H-binding protein, FHBP)、奈瑟菌粘附素A(Neisseria adhesin A, NadA)及奈瑟菌肝素结合抗原(Neisseria heparin-binding antigen, NHBA)等。正是由于采用了这种计算加实验的转化医学策略,目前,MenB疫苗已在上市审批中[84]。
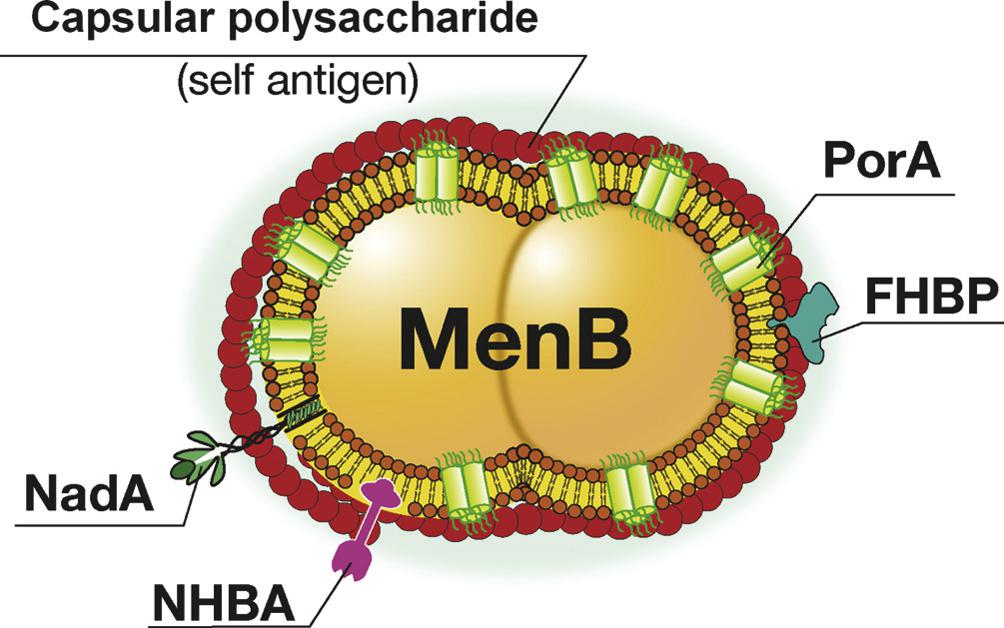
图7-9 脑膜炎奈瑟菌B示意图
5. 在移植免疫中的应用
人工神经网络等机器学习方法已经用于移植免疫领域多年;例如,用于预测肝移植排斥、肝移植后他克莫司血药浓度、辅助诊断肾移植超急排斥、评估肾移植并发症、预测肾移植后巨细胞病毒感染、预测胰腺移植的预后等。器官移植,最重要的是HLA配型。然而,由于供者有限,在没有完全匹配的情况下,如何从不完全匹配的供者选出最合适、最兼容的。美国匹兹堡大学医学中心的Duquesnoy教授研究该问题10余年,开发了一系列基于EXCEL的宏程序HLAMatchmaker[85]。由于该程序要创建临时文件并要反复地在EXCEL表格之间剪切粘贴,既费时间又容易出错,因此Filho等最近开发了基于HLAMatchmaker的EpHLA程序,实现的分析过程的完全自动化。[86]
6. 在变态反应防治中的应用
民以食为天。然而,自古以来食物过敏(food allergy)就一直困扰着人类。流传于民间和传统医学中的“发物”之说以及与之相关的种种饮食禁忌,在一定程度上反应了人们对食物过敏的经验认识。现代医学与免疫学研究表明,绝大多数食物过敏属于IgE介导的I型变态反应,一般表现为哮喘、腹泻、腹痛、荨麻疹等;但严重的也可能出现危及生命的、与青霉素过敏反应类似的过敏性休克。流行病学调查的数据显示,食物过敏在成年人中的发病率约为2 ~4%;而在儿童中则更高达8%;每年每100万人中,约32人发生严重甚至致命的食物过敏[87]。随着转基因农作物的出现及其在食品工业中的广泛应用,公众对食物过敏的担忧进一步加深。了解、获取食物过敏的相关信息已经成为公众的迫切需求。“新买的转基因玉米会不会有更高的引起食物过敏的风险?”、“我吃豌豆过敏,那吃山里朋友送来的土特产小扁豆会不会也过敏呢?”人们自然而然会有诸如此类的种种问题。也就是说,公众迫切需要一个与天气预报与预警类似的,使用方便、通俗易懂的食物过敏预测、预警与查询系统。
由于食物过敏极为常见,危害广泛,且与公众日常生活与健康息息相关,美国、日本、欧盟国家及相关国际组织如世界卫生组织(World Health Organization, WHO)、联合国粮农组织(Food and Agriculture Organization, FAO)一直非常重视食物过敏及其防治研究。现代医学研究认为,食物过敏主要由食物中一些特别的蛋白质引起;这些引起过敏(变态反应)的蛋白质在免疫学中被统称为变应原(allergen)。目前,对食物过敏除了一些对症治疗外,并没有根治措施。因此,避免接触或摄入过敏食物,从而预防食物过敏的发生就至关重要了。临床上,主要通过皮试和体外IgE检测来诊断食物过敏。然而,临床诊断用变应原的种类有限,而食物及其蛋白质组成却纷繁复杂。如何根据有限的实验结果,科学严谨地推断到更多未经实验检测的食品(包括转基因食品)中去,从而预测、预警其引起食物过敏的风险,这已经成为免疫信息学研究的一个前沿与热点。
这主要体现在以下两个方面:其一是1996年以来,大量变应原数据库及IgE表位数据库出现;其二是食物过敏预测方法日趋成熟。目前,食物过敏的免疫信息学预测已经发展出短肽匹配、序列比对、结构比较等3种不同方法[88]。短肽匹配方法比较用户提交的蛋白质氨基酸序列与数据库中所有已知过敏原有没有8个连续相同氨基酸。如果有,则认为该蛋白质可能引起食物过敏。在美国、欧盟、日本,短肽匹配方法已经广泛用于转基因植物的安全评估中。序列比对方法采用BLAST或FASTA程序,把用户提交的蛋白质氨基酸序列与数据库中所有已知过敏原进行序列相似性的两两比对。2001年,FAO/WHO专家组推荐,两比对序列80个氨基酸残基的序列节段内,如果35%以上的氨基酸残基相同则预测该蛋白质可能引起过敏反应。新近的研究显示,两条比对好的序列中如有70%以上的氨基酸残基相同,则几乎肯定会有食物过敏的发生。例如,Sanchez-Monge等报道,对豌豆过敏的18位患者同时也都对小扁豆过敏[89]。豌豆中的过敏原主要是豌豆种子球蛋白(vicilin)和伴球蛋白(convicilin);而小扁豆的种子球蛋白与豌豆种子球蛋白有90%以上的氨基酸残基相同,豌豆伴球蛋白与小扁豆伴球蛋白之间,相同氨基酸残基超过70%。Beyer等报道[90],14位对榛子过敏的患者中,12位经检测有能与11S榛子球蛋白结合的IgE。同时,这14位对榛子过敏的患者约有一半也对花生或核桃、巴西果、腰果、杏仁等过敏。这些坚果的11S球蛋白序列有45%~55%的氨基酸残基与榛子相同。本领域最新的研究思路是通过比较结构进行食物过敏预测。由于食物过敏主要由IgE介导,而大多数IgE识别的是变应原上的空间构象性表位;同时,大多数重要的变应原都可归结到少数几个结构家族,提示只要与已知变应原具有相似空间结构,即使氨基酸序列相似性程度低,也可能形成相似的空间构象性表位,并因此可能引发交叉的过敏反应。2005年,在西班牙召开了一场关于过敏预测方法的专题国际学术讨论会。与会专家一致认为, FAO/WHO专家组2001年推荐的双测试中,6连续氨基酸短肽匹配方法假阳性率高,不主张继续采用。对序列比对方法中的同率阈值,与会专家存在分歧,主流意见认为,FAO/WHO专家组2001年推荐的35%的阈值较为保守,但仍可在应用中继续检验。此外,与会专家还一致看好结构比较方法,但由于目前结构数据仍然缺乏,同时还没有与序列比对类似的统一清晰的结构相似性指标,结构比较方法仍有待进一步研究。
药物过敏是另一种最为常见的变态反应。最近,药物过敏有突破性发现[91]。至少有部分药物,如阿巴卡韦、卡马西平等导致严重甚至是致命的IV变态反应,其机制完全不同于传统观点。现已确定,阿巴卡韦与卡马西平能分别结合到HLA-B*57:01、HLA-B*15:02分子的抗原结合槽,从而使相应分子递呈抗原肽的特性发生改变,就仿佛用药后机体有了一个新的HLA分子,从而导致具有该HLA等位基因的患者发生类似器官移植不匹配的后果。今后,免疫信息学在预测药物过敏方面必定大有所为。免疫信息学还能用到什么地方?未来总是超乎想象。
脚注
* 诺华基金会是一个国际性的科学和教学慈善机构,它旨在促进生物学、医学和化学研究方面的合作。John Wiley从1986年起就是诺华基金会的出版商,出版了独一无二的、受到业界高度尊重的诺华系列丛书。这些书籍包括诺华基金研讨会的论文集,并且还汇编了主要科学家和学者演讲后广泛的鼓舞人心的讨论和辩论。这些珍贵的资源涵盖了20世纪后期所有关键的生物学发展,并且由国际知名的专家作为撰稿人,其中更包括很多诺贝尔奖获得者。生动的辩论加上撰稿人的国际地位,为诺华基金研讨会系列丛书赢得了在科学文献方面独一无二的尊贵地位。
# 1971年Anthony Nolan出生并发现患有Wiskott-Aldrich综合征,只能通过骨髓移植治疗,但当时没有寻找除至亲之外的供者系统。1973年,世界上第一例无亲缘关系的配型与骨髓移植成功。居住在澳洲的安东尼妈妈雪莉.诺南(Shirley Nolan)看到了希望,不远千里回到英国,呼吁国人踊跃验血,并于1975年催生了Anthony Nolan基金会,建立了世界上第一个骨髓资料库。可惜,安东尼.诺南没有等到合适的供者,于1979年去世。1993年安东尼.诺南研究所建成;迄今已有职员171位,登记的供者已超过40万份。
参考文献
Orosz CG: An introduction to immuno-ecology and immuno-informatics. In: Design Principles for the Immune System and Other Distributed Autonomous Systems. Edited by Segel LA, Cohen IR: Oxford University Press; 2001: 125–149.
Orosz CG: The case for immuno-informatics. Graft 2002, 5(8):462-465.
Segel LA: Controlling the immune system: Diffuse feedback via a diffuse informational network. In: Complexity in biological information processing: Novartis Foundation Symposium 239. Edited by Bock GR, Goode JA: John Wiley & Sons; 2001: 31–44.
Petrovsky N, Schonbach C, Brusic V: Bioinformatic strategies for better understanding of immune function. In Silico Biol 2003, 3(4):411-416.
Rammensee HG: Immunoinformatics: bioinformatic strategies for better understanding of immune function. Introduction. Novartis Found Symp 2003, 254:1-2.
Hart E, Timmis J: Application areas of AIS: The past, the present and the future. Applied Soft Computing 2008, 8(1):191-201.
Timmis J: Artificial immune systems-today and tomorrow. Natural Computing 2007, 6(1):1-18.
Flower DR, McSparron H, Blythe MJ, Zygouri C, Taylor D, Guan P, Wan S, Coveney PV, Walshe V, Borrow P et al: Computational vaccinology: quantitative approaches. Novartis Found Symp 2003, 254:102-120; discussion 120-105, 216-122, 250-102.
Pinheiro CS, Martins VP, Assis NR, Figueiredo BC, Morais SB, Azevedo V, Oliveira SC: Computational vaccinology: an important strategy to discover new potential S. mansoni vaccine candidates. J Biomed Biotechnol 2011, 2011:503068.
Rappuoli R, Covacci A: Reverse vaccinology and genomics. Science 2003, 302(5645):602.
Jones D: Reverse vaccinology on the cusp. Nat Rev Drug Discov 2012, 11(3):175-176.
Hagmann M: Computers aid vaccine design. Science 2000, 290(5489):80-82.
Kuroda D, Shirai H, Jacobson MP, Nakamura H: Computer-aided antibody design. Protein Eng Des Sel 2012:in press.
Tomar N, De RK: Immunoinformatics: an integrated scenario. Immunology 2010, 131(2):153-168.
Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW: GenBank. Nucleic Acids Res 2012, 40(Database issue):D48-53.
UniProtConsortium: Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res 2011, 39(Database issue):D214-219.
Madej T, Addess KJ, Fong JH, Geer LY, Geer RC, Lanczycki CJ, Liu C, Lu S, Marchler-Bauer A, Panchenko AR et al: MMDB: 3D structures and macromolecular interactions. Nucleic Acids Res 2012, 40(Database issue):D461-464.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M: KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012, 40(Database issue):D109-114.
Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J et al: IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res 2009, 37(Database issue):D1006-1012.
Salimi N, Fleri W, Peters B, Sette A: The Immune Epitope Database: A Historical Retrospective of the First Decade. Immunology 2012:in press.
Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE et al: Immune epitope database analysis resource. Nucleic Acids Res 2012, 40(Web Server issue):W525-530.
Robinson J, Mistry K, McWilliam H, Lopez R, Marsh SG: IPD--the Immuno Polymorphism Database. Nucleic Acids Res 2010, 38(Database issue):D863-869.
Johnson G, Wu TT: Kabat Database and its applications: future directions. Nucleic Acids Res 2001, 29(1):205-206.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S: SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999, 50(3-4):213-219.
Singh MK, Srivastava S, Raghava GP, Varshney GC: HaptenDB: a comprehensive database of haptens, carrier proteins and anti-hapten antibodies. Bioinformatics 2006, 22(2):253-255.
Sayers S, Ulysse G, Xiang Z, He Y: Vaxjo: a web-based vaccine adjuvant database and its application for analysis of vaccine adjuvants and their uses in vaccine development. J Biomed Biotechnol 2012, 2012:831486.
Saha S, Bhasin M, Raghava GP: Bcipep: a database of B-cell epitopes. BMC Genomics 2005, 6:79.
Huang J, Honda W: CED: a conformational epitope database. BMC Immunol 2006, 7:7.
Yang B, Sayers S, Xiang Z, He Y: Protegen: a web-based protective antigen database and analysis system. Nucleic Acids Res 2011, 39(Database issue):D1073-1078.
Jongeneel V: Towards a cancer immunome database. Cancer Immun 2001, 1:3.
Zhang ZH, Tan SC, Koh JL, Falus A, Brusic V: ALLERDB database and integrated bioinformatic tools for assessment of allergenicity and allergic cross-reactivity. Cell Immunol 2006, 244(2):90-96.
Mills EN, Valovirta E, Madsen C, Taylor SL, Vieths S, Anklam E, Baumgartner S, Koch P, Crevel RW, Frewer L: Information provision for allergic consumers--where are we going with food allergen labelling? Allergy 2004, 59(12):1262-1268.
Mills EN, Jenkins JA, Sancho AI, Miles S, Madsen C, Valovirta E, Frewer L: Food allergy information resources for consumers, industry and regulators. Arb Paul Ehrlich Inst Bundesamt Sera Impfstoffe Frankf A M 2006(95):17-25; discussion 25-17.
Ivanciuc O, Schein CH, Braun W: SDAP: database and computational tools for allergenic proteins. Nucleic Acids Res 2003, 31(1):359-362.
Allcorn LC, Martin AC: SACS--self-maintaining database of antibody crystal structure information. Bioinformatics 2002, 18(1):175-181.
Huang J, Kawashima S, Kanehisa M: New amino acid indices based on residue network topology. Genome Inform 2007, 18:152-161.
Huang J, Honda W, Kanehisa M: Predicting B cell epitope residues with network topology based amino acid indices. Genome Inform 2007, 19:40-49.
Larsen JE, Lund O, Nielsen M: Improved method for predicting linear B-cell epitopes. Immunome Res 2006, 2:2.
Zhang W, Xiong Y, Zhao M, Zou H, Ye X, Liu J: Prediction of conformational B-cell epitopes from 3D structures by random forests with a distance-based feature. BMC Bioinformatics 2011, 12:341.
Kulkarni-Kale U, Bhosle S, Kolaskar AS: CEP: a conformational epitope prediction server. Nucleic Acids Res 2005, 33(Web Server issue):W168-171.
Haste Andersen P, Nielsen M, Lund O: Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci 2006, 15(11):2558-2567.
Sun J, Wu D, Xu T, Wang X, Xu X, Tao L, Li YX, Cao ZW: SEPPA: a computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res 2009, 37(Web Server issue):W612-616.
Hu X, Zhou W, Udaka K, Mamitsuka H, Zhu S: MetaMHC: a meta approach to predict peptides binding to MHC molecules. Nucleic Acids Res 2010, 38(Web Server issue):W474-479.
Zhang GL, Ansari HR, Bradley P, Cawley GC, Hertz T, Hu X, Jojic N, Kim Y, Kohlbacher O, Lund O et al: Machine learning competition in immunology - Prediction of HLA class I binding peptides. J Immunol Methods 2011, 374(1-2):1-4.
Smith GP: Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228(4705):1315-1317.
Devlin JJ, Panganiban LC, Devlin PE: Random peptide libraries: a source of specific protein binding molecules. Science 1990, 249(4967):404-406.
Smith GP, Petrenko VA: Phage Display. Chem Rev 1997, 97(2):391-410.
Huang J, Gutteridge A, Honda W, Kanehisa M: MIMOX: a web tool for phage display based epitope mapping. BMC Bioinformatics 2006, 7:451.
Huang J, Ru B, Dai P: Prediction of protein interaction sites using mimotope analysis. In: Protein-Protein Interactions - Computational and Experimental Tools. Edited by Cai W: InTech; 2012: 189-206.
Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista M, Ferracuti S, Nelson B, Paoluzi S et al: A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 2002, 295(5553):321-324.
Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, Monk P, Shorrock-Hart CP, Jermutus L, Minter RR: Probing a protein-protein interaction by in vitro evolution. Proc Natl Acad Sci U S A 2006, 103(20):7619-7624.
Pasqualini R, Ruoslahti E: Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380(6572):364-366.
Hsiung PL, Hardy J, Friedland S, Soetikno R, Du CB, Wu AP, Sahbaie P, Crawford JM, Lowe AW, Contag CH et al: Detection of colonic dysplasia in vivo using a targeted heptapeptide and confocal microendoscopy. Nat Med 2008, 14(4):454-458.
Macdougall IC, Rossert J, Casadevall N, Stead RB, Duliege AM, Froissart M, Eckardt KU: A peptide-based erythropoietin-receptor agonist for pure red-cell aplasia. N Engl J Med 2009, 361(19):1848-1855.
Knittelfelder R, Riemer AB, Jensen-Jarolim E: Mimotope vaccination--from allergy to cancer. Expert Opin Biol Ther 2009, 9(4):493-506.
Lee YJ, Yi H, Kim WJ, Kang K, Yun DS, Strano MS, Ceder G, Belcher AM: Fabricating genetically engineered high-power lithium-ion batteries using multiple virus genes. Science 2009, 324(5930):1051-1055.
Nam YS, Magyar AP, Lee D, Kim JW, Yun DS, Park H, Pollom TS, Jr., Weitz DA, Belcher AM: Biologically templated photocatalytic nanostructures for sustained light-driven water oxidation. Nat Nanotechnol 2010, 5(5):340-344.
Smothers JF, Henikoff S, Carter P: Affinity selection from biological libraries. Science 2002, 298(5593):621-622.
Menendez A, Scott JK: The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal Biochem 2005, 336(2):145-157.
Vodnik M, Zager U, Strukelj B, Lunder M: Phage display: selecting straws instead of a needle from a haystack. Molecules 2011, 16(1):790-817.
Thomas WD, Golomb M, Smith GP: Corruption of phage display libraries by target-unrelated clones: diagnosis and countermeasures. Anal Biochem 2010, 407(2):237-240.
Brammer LA, Bolduc B, Kass JL, Felice KM, Noren CJ, Hall MF: A target-unrelated peptide in an M13 phage display library traced to an advantageous mutation in the gene II ribosome-binding site. Anal Biochem 2008, 373(1):88-98.
Derda R, Tang SK, Li SC, Ng S, Matochko W, Jafari MR: Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification. Molecules 2011, 16(2):1776-1803.
Ru B, Huang J, Dai P, Li S, Xia Z, Ding H, Lin H, Guo F, Wang X: MimoDB: a New Repository for Mimotope Data Derived from Phage Display Technology. Molecules 2010, 15(11):8279-8288.
Huang J, Ru B, Zhu P, Nie F, Yang J, Wang X, Dai P, Lin H, Guo FB, Rao N: MimoDB 2.0: a mimotope database and beyond. Nucleic Acids Res 2012, 40(Database issue):D271-277.
Huang J, Ru B, Dai P: Bioinformatics resources and tools for phage display. Molecules 2011, 16(1):694-709.
Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ: RELIC--a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics 2004, 4(5):1439-1460.
Huang J, Ru B, Li S, Lin H, Guo FB: SAROTUP: scanner and reporter of target-unrelated peptides. J Biomed Biotechnol 2010, 2010:101932.
Abhinandan KR, Martin AC: Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol 2008, 45(14):3832-3839.
Deret S, Maissiat C, Aucouturier P, Chomilier J: SUBIM: a program for analysing the Kabat database and determining the variability subgroup of a new immunoglobulin sequence. Comput Appl Biosci 1995, 11(4):435-439.
Martin AC: Accessing the Kabat antibody sequence database by computer. Proteins 1996, 25(1):130-133.
Abhinandan KR, Martin AC: Analyzing the "degree of humanness" of antibody sequences. J Mol Biol 2007, 369(3):852-862.
Abhinandan KR, Martin AC: Analysis and prediction of VH/VL packing in antibodies. Protein Eng Des Sel 2010, 23(9):689-697.
Whitelegg NR, Rees AR: WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng 2000, 13(12):819-824.
Arnold K, Bordoli L, Kopp J, Schwede T: The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22(2):195-201.
Marcatili P, Rosi A, Tramontano A: PIGS: automatic prediction of antibody structures. Bioinformatics 2008, 24(17):1953-1954.
Sircar A, Kim ET, Gray JJ: RosettaAntibody: antibody variable region homology modeling server. Nucleic Acids Res 2009, 37(Web Server issue):W474-479.
Tseng YY, Dupree C, Chen ZJ, Li WH: SplitPocket: identification of protein functional surfaces and characterization of their spatial patterns. Nucleic Acids Res 2009, 37(Web Server issue):W384-389.
Liang J, Edelsbrunner H, Woodward C: Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci 1998, 7(9):1884-1897.
Xiang Z, Todd T, Ku KP, Kovacic BL, Larson CB, Chen F, Hodges AP, Tian Y, Olenzek EA, Zhao B et al: VIOLIN: vaccine investigation and online information network. Nucleic Acids Res 2008, 36(Database issue):D923-928.
He Y, Xiang Z, Mobley HL: Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol 2010, 2010:297505.
Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ et al: Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 2000, 287(5459):1809-1815.
Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, Jennings GT, Baldi L, Bartolini E, Capecchi B et al: Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287(5459):1816-1820.
Black S, Pizza M, Nissum M, Rappuoli R: Toward a meningitis-free world. Sci Transl Med 2012, 4(123):123ps125.
Duquesnoy RJ: Antibody-reactive epitope determination with HLAMatchmaker and its clinical applications. Tissue Antigens 2011, 77(6):525-534.
Filho HL, da Mata Sousa LC, von Glehn Cde Q, da Silva AS, dos Santos Neto Pde A, do Nascimento F, de Castro AF, do Nascimento LM, Kneib C, Bianchi Cazarote H et al: EpHLA software: a timesaving and accurate tool for improving identification of acceptable mismatches for clinical purposes. Transpl Immunol 2012, 26(4):230-234.
Gibson J: Bioinformatics of protein allergenicity. Mol Nutr Food Res 2006, 50(7):591.
Goodman RE: Practical and predictive bioinformatics methods for the identification of potentially cross-reactive protein matches. Mol Nutr Food Res 2006, 50(7):655-660.
Sanchez-Monge R, Lopez-Torrejon G, Pascual CY, Varela J, Martin-Esteban M, Salcedo G: Vicilin and convicilin are potential major allergens from pea. Clin Exp Allergy 2004, 34(11):1747-1753.
Beyer K, Grishina G, Bardina L, Grishin A, Sampson HA: Identification of an 11S globulin as a major hazelnut food allergen in hazelnut-induced systemic reactions. J Allergy Clin Immunol 2002, 110(3):517-523.
Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA et al: Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486(7404):554-558.